We are preparing new PreMetabo web site.
As soon as possible, we will release the web based PreMetabo service.
We are preparing new PreMetabo web site.
As soon as possible, we will release the web based PreMetabo service.
Carcinogenicity is a toxicity that causes cancer in body. Generally carcinogenicity test requires long time (usually 2 years), currently only in vivo test methods are established. Usually the test uses mice or rats, exposing them to a compound. And the variable to be observed is existence of cancer. PreADMET predicts the result from its model, which is built from the data of NTP (National Toxicology Program) and US FDA, which are the results of the in vivo carcinogenicity tests of mice and rats for 2 years.
| Type | NTP Definition | Description |
| negative | Clear evidence of carcinogenic activity | negative prediction |
| positive | No evidence of carcinogenic activity | positive prediction |
Ames test is a simple method to test mutagenicity of a compound, which is suggested by Dr. Ames. It uses several strains of the bacterium Salmonella typhimurium that carry mutations in genes involved in histidine synthesis, so that they require histidine for growth. The variable being tested is the mutagen’s ability to cause a reversion to growth on a histidine-free medium. [Bruce N. Ames, E. G. Gurney, James A. Miller, and H. Bartsch (1973). “Carcinogens as Frameshift Mutagens: Metabolites and Derivatives of 2-acetylaminofluorene and other Aromatic Amine Carcinogens”. PNAS 69: 3128-213] [ Ames,B.N. et al. PNAS. 1972, 69, 3128. ]
PreADMET predicts toxicity to TA98, TA100 and TA1535 which are often used in Ames test. And the result can be calculated both with consideration of metabolite (Metabolic activation by rat liver 10% homogenate, +S9) and without consideration of metabolite. (No metabolic activation, -S9) The actual value of the prediction result is “positive” or “negative”.
|
Type |
NTP Definition | Description |
|
negative |
no change of population (vs. blank plate) |
negative prediction |
| positive | change of population, more than double of blank plate’s change |
positive prediction |
This measurement is based on compounds that have molecular properties within the 90 % upper bound found in the WDI (World Drug Index).
[ Brown,R.D. et al. ”Tools for designing diverse, drug-like, cost-effective combinatorial libraries”; in Combinatorial Library Design and Evaluation”, Marcel Dekker, Inc. New York, 2001, 328. ]
| Descriptors | 90% cutoff | Descriptors | 90% cutoff |
| MW | 550 | Kappa-2A | 12 |
| Rotbond | 13 | Kappa-3A | 8 |
| Hbond acceptor | 9 | CHI-V-0 | 20 |
| Hbond donor | 5 | CHI-V-1 | 12 |
| AlogP | 5 | CHI-V-2 | 10 |
| MolRef | 120 | CHI-V-3P | 8 |
| Balaban Jx | 2.8 | CHI-V-3C | 2.2 |
| PHI | 8 | Wiener | 4000 |
| Kappa-1A | 25 | Zabreb | 175 |
Filtering by reactive functional group is the one of the important steps for the drug discovery. A compound with a reactive functional group is reactive in human body, and it has possibility to cause toxicity. Additionally, a compound with a reactive functional group leads to reaction during a process of an in vitro experiment, such as a case of HTS. This may results some serious error of activity for drug.
These compounds can be filtered off or removed, at the beginning of drug discovery. This filtering reduces probability of causing toxicity or error. PreADMET can find compounds with electrophilic functional group which tends to make a covalent bond with protein, causing false positive. And PreADMET also finds compounds with 3 prohibited functional groups that are extremely unusual in drug discovery. The number of both kinds of functional groups are 23 in total.
[ (1) Rishton,G.M. DDT. 1997, 2, 382. (2) Walters,W.P. et al. Adv. Drug Deliv. Rev. 2002, 54, 255. ]
MDDR-like rule is published by Tudor I. Oprea. The rule of five test produced similar results when applied to the ACDF and MDDRF subset, which 80% of ACDF and MDDRF pass the rule of five test. ACD database is non-drug database and MDDR database is drug database. ACDF and MDDRF are databases removed the reactive functional groups such as acyl-halides, sulfonyl-halides, Michael acceptors, etc.. In this study, therefore, he has concluded that the rule of five test cannot be used to discriminate between drugs and non-drugs. Descriptors used to MDDR-like rule are the number of rings, the number of rigid bonds and the number of rotatable bonds.
The probability of finding a ‘druglike’ compound is higher in its ranges (e.g., No.Rings ≥ 3, No. Rigid bonds ≥ 18, No. Rotatable bonds ≥ 6), while the probability of finding a ‘nondrug-like’ compound is higher in the ranges (e.g., No.Rings ≤ 2, No. Rigid bonds ≤ 17, No. Rotatable bonds ≤ 5). There is more information in the table below. In PreADMET, The result from MDDR-like rule is represented as classification of the following table and this rule can be customized by user. (Figure 5.5)
[ Oprea,T.I. J. Comput. Aid. Mol. Des. 2000, 14, 251. ]
| Condition | |
| drug-like | No. Rings ≥ 3
No. Rigid bonds ≥ 18 No. Rotatable bonds ≥ 6 |
| nondrug-like | No. Rings ≤ 2
No. Rigid bonds ≤ 17 No. Rotatable bonds ≤ 5 |
| mid-structure | structures of the other ranges |
CMC-like rule is similar to rule of five. This is published by Arup K. Ghose et al.. They defined druglike character for the CMC database, which is removed several classes of compounds such as diagnostic imaging agents, solvents, and pharmaceutical aids. In this study, the qualifying range (covering more than 80% of the compounds) of the calculated log P is between -0.4 and 5.6, with an average value of 2.52. For molecular weight, the qualifying range is between 160 and 480, with an average value of 357. For molar refractivity, the qualifying range is between 40 and 130, with an average value of 97. For the total number of atoms, the qualifying range is between 20 and 70, with an average value of 48. The following table is about the qualifying range.
[ Ghose,A.K. et al. J. Comb. Chem. 1999, 1, 55. ]
| drug class | Qualifying Range in CMC Database | ||||
| AlogP
(80%) |
AMR
(80%) |
Molecular Weight
(80%) |
Number of Atoms
(80%) |
||
| CMC clean | -0.4 ~ 5.6 | 40 ~ 130 | 160 ~ 480 | 20 ~ 70 | |
| inflammatory | 1.4 ~4.5 | 59 ~ 119 | 212 ~ 447 | 24 ~ 59 | |
| depressant | 1.4 ~ 4.9 | 62 ~ 114 | 210 ~ 380 | 32 ~ 56 | |
| psychotic | 2.3 ~ 5.2 | 85 ~ 131 | 274 ~ 464 | 40 ~ 63 | |
| hypertensive | -0.5 ~ 4.5 | 54 ~ 128 | 206 ~ 506 | 28 ~ 66 | |
| hypnotic | 0.5 ~ 3.9 | 43 ~ 97 | 162 ~ 360 | 20 ~ 45 | |
| neoplastic | -1.5 ~ 4.7 | 43 ~ 128 | 180 ~ 475 | 21 ~ 63 | |
| infective | -0.3 ~ 5.1 | 44 ~ 144 | 145 ~ 455 | 12 ~ 64 | |
Identification and optimization of lead compounds as chemical staring points are very important in combinatorial chemistry. Lead-like rule is published by Simon J. Teague et al. and defined to consider designing libraries with druglike physicochemical properties. They have divided the common sources of lead compounds for drug discovery into three types like the following figure. [ Teague,S.J.et al., Angew. Chem. Int. Ed. 1999, 38, 3743. ]
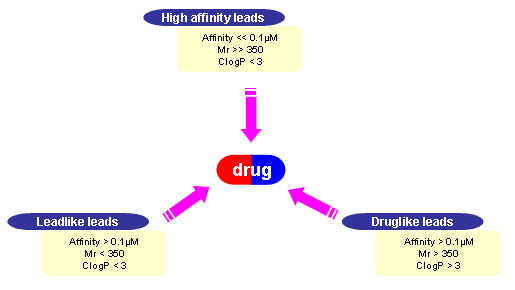
Figure. Group of Leads of Drug
Lipinski’s Rule, so called “Rule of Five”, is published by Christopher A. Lipinski et al. in Pfizer Central Research (Groton, NJ,USA). They selected a subset of 2245 compounds from WDI (World Drug Index) database and defined drug-like character through this subset. The results are the followings: [ Lipinski,C.A. et al. Adv. Drug Deliv. Rev. 1997, 23, 3. ]
About 90 % of the above subset is included in defined rage with better solubility and permeability.
Generally, only the unbound drug is available for diffusion or transport across cell membranes, and also for interaction with a pharmacological target. As a result, a degree of plasma protein binding of a drug influences not only on the drug’s action but also its disposition and efficacy. In PreADMET can predict percent drug bound in plasma protein as in vitro data on human.
Although there are some differences in the experimental values by compounds or their metabolisms, we can put into general categories like below.
| Classification | Plasma Protein Binding(%PPB) |
| Chemicals strongly bound | more than 90% |
| Chemicals weakly bound | less than 90% |